Using DRAM2: for DRAM1 Users
Getting started with DRAM2, for those who are most familiar with DRAM1.
Welcome to Members of the Wrighton Lab!
This document is meant for you only. You as a lab member are privy to the information on DRAM2 before anyone else. Please take care to note any concerns you have with this document, as it will be used to make a generalized version for the public. The world will be very grateful for the time you take to look over this document!
The Point of This Document
As you know, we are migrating to DRAM2, which should be an improvement on DRAM1. Documentation will be different for new users as compared to those who have already used DRAM1. This document is intended to get DRAM1 users in the Wrighton lab ready to use DRAM2, and will focus mostly on differences and new features, and less on how it works fundamentally.
As you look over this document, notice that DRAM2 differs from DRAM1 in these key ways:
It has a project config that can store run data. This is used for checking, and lets us skip most arguments.
It has more steps than DRAM1, but each step is simpler.
DRAM-v is not part of DRAM2. It may be in the future, but not currently.
There is only one command, dram2 which lets you use all sub commands.
Only DRAM2 has Adjectives and Phylogenetic Trees.
Not everything from DRAM1 is implemented in DRAM2 yet.
Preview
Before we start, I want to lay out a simple example. As with the points above, it would be good to keep this in the back of your mind while you read the rest of this document.
source /opt/Miniconda2/miniconda2/bin/activate DRAM1.4.4
DRAM.py annotate \
-i "./input_fasta_files/*.fasta" \
-o soil/dram1_out \
--use_camper --use_fegenie --use_sulfur
Is the same as:
source /opt/Miniconda2/miniconda2/bin/activate DRAM2BETA
dram2 -d from_fasta -t 2 call \
./input_fasta_files/*.fasta
dram2 -d from_fasta -t 30 annotate \
--use_dbset mini_kegg
Grab an Example Directory
The 15 soil genomes used in the DRAM1 paper are a good place to start. We could call all the soil genomes, but that would take too long, even for this limited example. Calling genes, and some other tools, are now using multiprocessing, but there is still a lot more work to do. So I selected two of those genomes and put them in a directory along with the steps complete so you can jump in.
Just copy the directory into a good working location on zenith.
cp -r /home/projects-wrighton-2/DRAM/development_flynn/dram2_dev/jan_26_23_main_pipeline/example_files_try_one ./my_dram_test_dir
cd ./my_dram_test_dir
It contains these folders
Activate the Environment
First, let’s activate the DRAM2 environment. This is just like with DRAM1. The first version of DRAM2 is DRAM2.0.rc1. This name indicates that it is DRAM major release 2, minor release 0, or the first, and then it is a release candidate 1. This test release will have that name in the output but be named DRAM2BETA because we are not yet at the release stage. Run the command below, and we can get started testing this release candidate.
source /opt/Miniconda2/miniconda2/bin/activate DRAM2BETA
Just a quick note on the setup. The DRAM2 config does not live with DRAM2. The
global config on zenith is at /etc/dram_config.yaml. You as a user can make a
file in your home directories config file ~/.config/dram_config.yaml and that
will replace the global one for you only. We are not going to get into this much,
but you should know because that is a big change.
DRAM2 Command Structure
The DRAM2 command line structure is hierarchical in that DRAM2 has one main command, aka dram2 and no matter what you want to do, it starts with dram2, as opposed to DRAM.py and DRAM-setup.py. You provide general/universal options to dram2, and then specific options to the DRAM2 sub commands.
It is, in this specific case, necessary to draw a distinction between command line options and command line arguments. For our purposes, options are anything specified by a flag like --do_this or --use_this <value>, and arguments are positional at the end of a command.
Thus, the structure of a typical DRAM2 command is:
dram2 <general_options> <sub_command> <sub_command_options> <arguments>
This should become clearer as we go through the help.
Explore the Help
With any new program, it is good to explore the help. The DRAM2 help is a lot larger compared to DRAM1 and really needs a good checking in order to validate.
First, let’s look at the overview:
dram2 --help
Thus far, adjectives are the most refined form of DRAM output. At the end of this process, we want to be able to have adjectives generated, but the adjectives in DRAM2 have more requirements that need to be met first. Adjectives use several new features of DRAM2, such as database checking and phylogenetic trees. To learn more about adjectives and see what specifically is required, you can run:
dram2 adjectives --help
dram2 adjectives eval --help
The first step to any DRAM project is probably calling genes.
Call Genes, and Start a Project
In DRAM1, calling genes was part of the annotation process, but now it is done with the call command, and the annotation process only works on already-called genes. This adds a step but makes the process a lot simpler for a layperson to follow.
First, please read the help and make sure you understand it. We will reiterate some of what it says in the next section, however.
dram2 call --help
In the past, DRAM confused people by having them pass a string to call genes with a python command, so now we let bash handle this. This should be safer and result in fewer errors.
dram2 -d from_fasta -t 2 call \
./input_fasta_files/*.fasta
Recall the notes about commands above.
Notice the output is specified by a -o and is passed to the dram2 command before the call command runs, the same with the -t command that tells dram the most cores it needs are 2.
The commands that get passed to dram2 are universal and work with all dram2 sub-commands, but you don’t pass them after the sub-commands.
So dram2 call -d would not work. The reverse is also true: you don’t pass an option to dram2 that goes to the sub-commands, so dram2 --prodigal_mode train call -0 soil/test1 would not work.
Additionally, dram2 call has a list of arguments after all the options for FASTAs. In DRAM1, the wild card path to FASTA files had to be a string. That was ok, but it was confusing at times. DRAM2 uses a normal file path instead.
If you have FASTAs that can’t be referenced with a regular expression, you can just add the paths one after another:
dram2 -vv -d soil/test1 -t 2 call \
./input_fasta_files/Cytophaga_hutchinsonii_ATCC_33406.fasta \
./input_fasta_files/Dechloromonas_aromatica_RCB.fasta
Note: multi-processing is on the FASTA level, so only two cores are needed.
Calling Annotations
Take a look at the help.
dram2 annotate --help
To use the FASTAs we just called and annotate them with all the databases that we need for adjectives, you can use this command. Note: you don’t need to point to the called genes so long as you use the same output directory.
dram2 -d from_fasta -t 30 annotate --use_dbset adjectives_kegg
To demonstrate a phylogenetic NXR-NAR tree, we need to have some specific genes in this example. Let’s annotate some modified FASTAs that have these genes added. You can annotate these called genes with the command:
dram2 -d called_annotated -t 30 annotate --use_dbset adjectives_kegg ./input_faa_files/*
Calling annotations can be done with a db_set, as seen above, but it can also be done with the --use_db flag individually. The one above would take a long time, but you can use these smaller databases to get a taste of annotations.
dram2 -d called_annotated -t 30 annotate --use_db fegenie --use_db camper --use_db methyl --use_db cant_hyd
There are also some databases that you may not think of as databases, such as Heme Motif count and even the genome statistics. It simply made sense to implement these as databases.
To see what sets/databases are available, you can use the help message, and you can learn more about the databases with this command:
This will have more information in the future.
Distillation
Distillation has not changed much compared to annotations. There are some exciting changes that will happen someday, but distillation is not yet the focus.
You will find that you can now select to only run some parts of the distillate. The output will include the CAMPER and Methyl sheets if annotations contain CAMPER or Methyl ids, and distill is now integrated with the history checker.
dram2 -d called_annotated distill
A Side Note On History Checks
If you try to run one of the dram2 annotate commands again, it will error by design; you have already annotated with these databases, so DRAM2 will not let you waste time or make a mistake by redoing them. You can still do so by using the force flag (-f).
dram2 -d called_annotated -t 30 annotate --use_db methyl
dram2 -d called_annotated -t 30 annotate -f --use_db methyl
If you call the genes for a FASTA but do not annotate it with the required databases, distill will give you an error, informing you of exactly what you are missing. The phylotree and adjectives commands will do the same. The force flag will once again allow you to continue, however ill-advised.
Phylogenetic Trees
Phylogenetic trees are more or less completely unique to DRAM2 and are used to determine the function of ambiguous genes using phylogeny. Currently, only the NXR/NAR tree is ready for this tool.

For our purposes here, we can simplify the process of this tool to a basic summary. The idea is that for each phylogenetic tree configured, this tool will:
Load in the pre-labeled tree and list of associated gene ids.
Filter genes to those needing clarification.
Label genes that fall into clades that all share the same label.
Label additional genes based on proximity.
dram2 -d called_annotated phylotree
This process depends on Annotation, and Adjectives now depend on this process. Unfortunately, we only have the NXR/NAR tree available in this test, but AMOA/PMOA is coming soon
A much more detailed outline of this system is in the works.
Adjectives Getting More Powerful
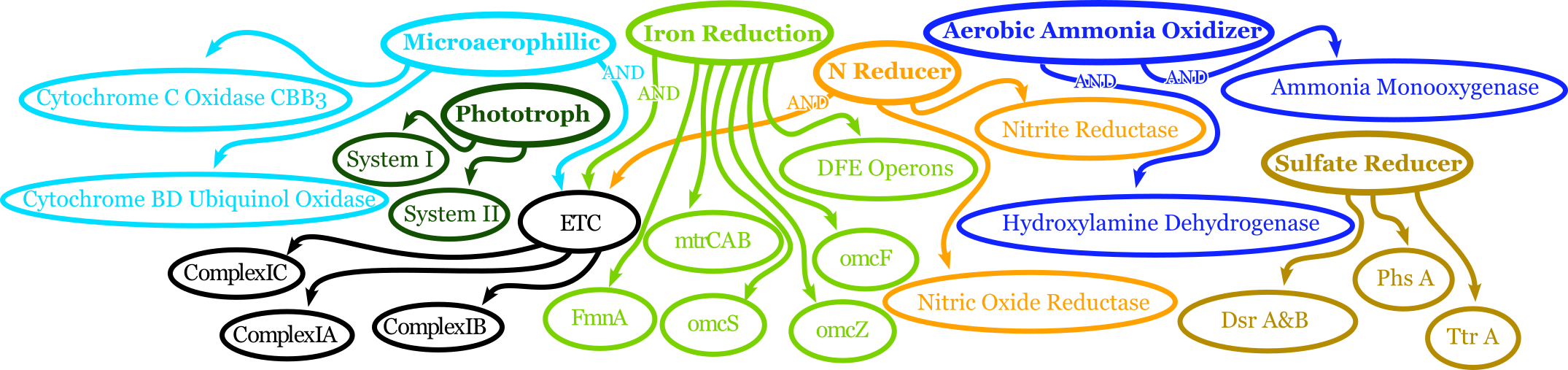
Adjectives are arriving in full force in DRAM2. The adjectives use a series of complex rules to ascribe attributes to genes. The rules in question include everything we have run in DRAM2 thus far. Once again, detailed documentation is coming in the near future. The figure above shows a very simplified view of how rule-based Genome Adjectives are assigned based on key systems. The true process has many more functions.
dram2 -d called_annotated adjectives eval
A Side Note on Verboseness
Many would not know about the -v AKA verbose option in DRAM1 because it made little difference. In DRAM2 we were able to attatch this option to the logging feature and give it a significant upgrade. The level is determined by the number of v’s passed to the dram2 command. There are 5 levels of verbosity which map onto the logging levels: 1=Critical, 2=Error, 3=Warning, 4=Info, 5=Debug. 5/Debug is the most informative, and 1/Critical only tells you the most serious errors.
You will learn more about how DRAM2 works and what is left to do with information in this annotation run.
dram2 -d called_annotated -t 30 -vvvvv annotate --use_db dbcan
This adjective run is nice and quiet.
dram2 -d called_annotated -v adjectives eval